Duchenne Muscular Dystrophy (Dmd) is a genetic disorder affecting 1 in 3,500 male births. It is caused by a mutation in the dystrophin gene, one of the longest genes in the human genome. Dystrophin is involved in cellular signaling and in the transmission of the force generated during muscle activity. Defective dystrophin proteins lead to muscle damage and failure. The nematode C. elegans uses dystrophin in much the same way humans do. As such, these tiny worms afford us the opportunity to study DMD with the unparalleled experimental ease associated with the study of these organisms.
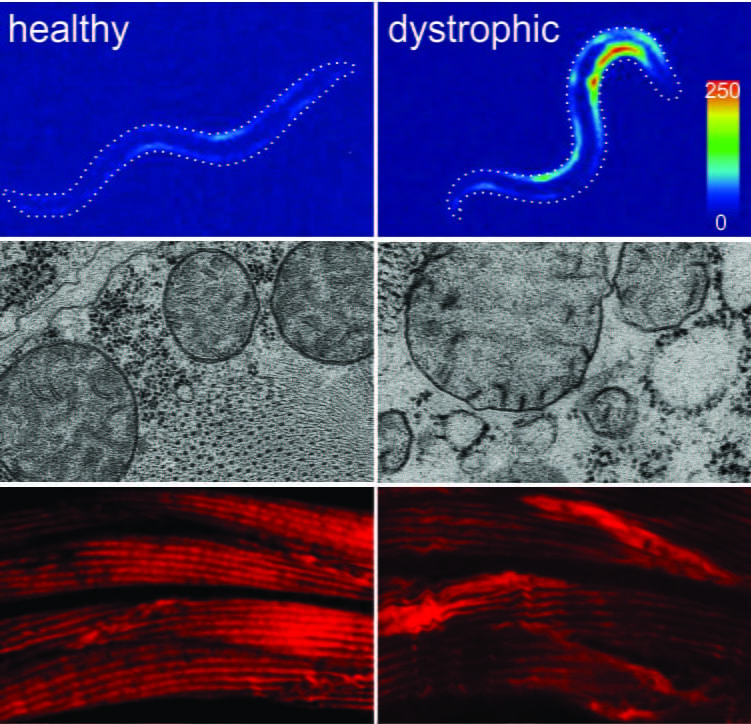
Figure 1. The musculature of healthy (left) and DMD worms (right) after burrowing in agar pipettes for a few days. heat map showing an excess of cytosolic calcium in dystrophic muscles (top) lis followed by loss of muscle mitochondria integrity (midle), and damage to the contractile machinery of the muscles (bottom). Click on the picture to read more.

Our lab uses a two-pronged in vivo/in vitro approach to study Dmd. The in vivo part of our approach uses nematodes modeling Dmd. We developed a high throughput automated assay that harnesses the natural burrowing behavior of nematodes to challenge motor output in healthy and dystrophic animals. Mutants mimicking Duchenne muscular dystrophy by lacking a functional ortholog of the dystrophin protein, DYS-1, crawl normally but are severely impaired in this assay. While healthy animals undergo healthy muscle hypertrophic growth, muscular degeneration in dystrophic (dys-1) mutants is hastened and exacerbated by burrowing. Dystrophic changes can be studied using a wide array of techniques including EM microscopy, calcium ratiometry, in depth automated kinematic analysis, confocal microscopy, immunohistochemistry, qPCR, and many more. To validate findings derived using nematode models of Dmd we use animal controls where the human dystrophin gene (hDMD) is used to rescue mutations in the nematode dystrophin gene (dys-1).
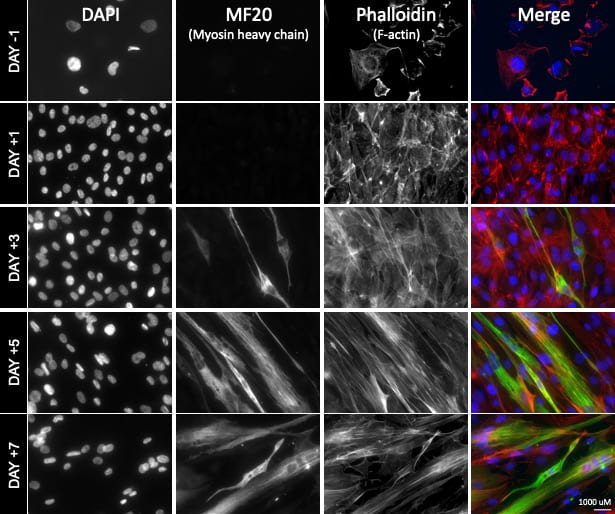
The second (in vitro) part of our approach involves the use of human muscle cell cultures (LHCN-M2) to study Dmd. Cell culture studies allow us to rapidly identify and evaluate potential treatment targets/compounds, and to accurately model mutations underlying Duchenne muscular dystrophy in different groups of patients. Our two approaches are synergistic and allow us to: A) provide a rapid and inexpensive human point of validation for nematode-derived insights, and B) to harness the in vivo, fast, and inexpensive nematode system for in vitro evaluation of human cell-culture derived insights.
In our lab, work on muscular dystrophy focuses on several goals:
1) Identifying the mechanisms by which muscles become impaired during the progression of Dmd.
2) Identifying cellular pathways able to stall the progression of Dmd in dystrophic muscles
3) Generating patient-specific in vivo and in vitro models of Dmd to evaluate potential treatment options
A dedicated team of researchers in our lab are working to improve the quality of life of individuals struggling with this terrible disease.








Our Duchenne muscular dystrophy team is comprised of Jessica Adams (Postdoc), Parham Jazireian (PhD), Danny Marchiafava (PhD), Adina Fazyl (MS), Shifat Niha (MS), Erinda Aidoo (URG), Annemarie Hyer (URG), Sabrina Kollbaum (URG).